|
Electronic Structure Methods for InterfacesThe interfaces we study consist of two parts: finite molecules - objects of conventional chemistry, and periodic solids - objects of conventional condensed matter physics. Accordingly, a good electronic structure method for interfaces should work well for both components, and more importantly, any interactions between them.
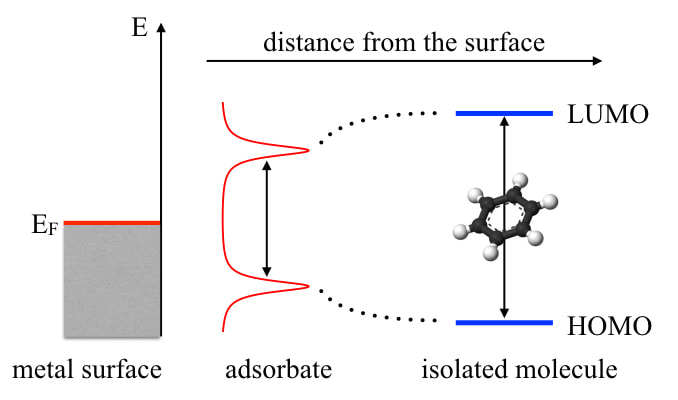
In the toughest case, the molecular subsystem could be strongly correlated in the chemistry sense (call that "static" if you so wish); the solid subsystem could be strongly correlated in the physics sense (call that "Hubbard U" if you so wish); and the interaction between them could involve non-negligible long-range Coulomb screening that is responsible for molecular level renormalization as we show in the left panel. There are few methods that can handle the above problems satisfactorily. Even when they do, chances are that they are so expensive that routine ab initio calculations of interfaces (which usually involve hundreds of atoms) are not realistic, at least for now. Our group attempts to develop new practical approaches for large-scale accurate interface electronic structure calculations. Of course, the methods we develop won't be the panacea for everything. Here, we focus on the property of level alignments - relative positions between molecular frontier orbitals and Fermi level of the metal or band edges of the semiconductor. It is important because of its relevance to interfacial dynamics such as charge transfer, especially in energy conversion applications. Currently, we hope to advance this field from three routes:
|
